パルスフィールド電気泳動法
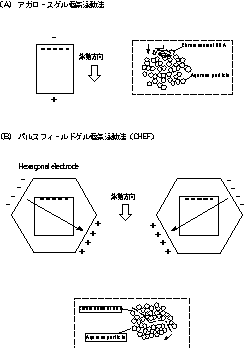
染色体の形態や数は、種によって特徴的で核型と呼ばれる。それを比較する方法は核型解析と呼ばれ、高等真核生物では分類や系統進化を調べる重要な方法として用いられている。しかしながら、酵母などの下等菌類では染色体の凝集像を見ることが困難なため、核型解析を行うことができなかった。近年、染色体DNAを分離する方法として開発されたパルスフィールド電気泳動法(PFGE)は、染色体をその大きさによってアガロースゲル上で分離する方法で、これにより調べられる核型は電気泳動核型と呼ばれる。染色体サイズが動植物のように大きくない酵母では、染色体を制限酵素で処理することなしにアガロースゲル中でバンドとして検出することが可能である。
<測定原理>
従来のアガロース電気泳動法は一方向から電場をかけてDNA分子をアガロースの網目の中で、その大きさに応じて分離するが、大きなDNA(数十kb以上)はアガロースの網目に引っかかって、分離することができなかった。これに対し、パルスフィールド電気泳動法は電場の方向を一定時間毎に変化させることによりDNA分子の形態的変化を誘発し、より巨大な数Mb以上のDNA分子をアガロースの網目の中をうまくすり抜けさせるように移動させ分離する方法である(図1)。サンプルの調製にあたっては染色体DNAを切らずに、本来の大きさを検出するために、操作過程での染色体DNAの分断や分解を避けなければならない。そのために、酵母細胞を低融点アガロース内に包埋し、その状態で細胞壁分解や細胞膜破壊や核タンパク質の分解など、全ての操作を行う。いったんアガロース内に包埋された染色体DNAは物理的な切断、分解から保護されているため、パルスフィールド電気泳動法のサンプルとして用いる事ができる。
<プロトコール>
1-1. パルスフィールド電気泳動用のサンプルの調製
このステップでは、酵母を低融点アガロースに包埋し、ザイモリエース(Zymolyase 20T)による細胞壁分解、SDS (Sodium dodecyl sulphate)による細胞膜や核膜の破壊、そしてプロテアーゼK (Proteinase K) によるタンパク質の分解を行う。酵母を包埋したアガロースは、染色体電気泳動装置(パルサーフォーシステムあるいはCHEF-DRIIシステム)に付属している型を用いて作製する。これらの型は各装置のコームの大きさにあったアガロースブロックを作成できる用に設計されている。
準備:
YPD液体培地(1% Yeast Extract、1% Bactopeptone、2% Glucose)
0.5M EDTA (pH7.5)
0.5M EDTA (pH9.0)
1M Tris-HCl (pH7.5)
Zymolyase 20T:5mg / ml in SCE
低融点アガロース:1%アガロース L(和光純薬)in 0.125M EDTA (pH7.5)
SCE:1M Sorbitol、0.1M Sodium Citrate (pH5.8)、10mM EDTA (pH8.0)
LET:0.5M EDTA (pH7.5)、10mM Tris-HCl (pH7.5)、7.5% 2-mercaptoehanol (2-ME)
NDS:0.5M EDTA (pH9.0)、10mM Tris-HCl (pH8.3)、0.06% SDS、0.17mg/ ml Proteinase K
方法:
注意:
*1 培養の時間、温度は目的に応じて変更可能。 *2 この時の細胞の濃度が重要である。目安としてはOD600が1.0程度が望ましい。あまり濃いと細胞壁分解の不十分な細胞が生じ、そういったのものでは細胞質中のDNA分解酵素がプロテアーゼKによる処理で失活しないため、アガロース・ブロック内で徐々に染色体DNAを分解することになり、電気泳動時におけるスメアーなバンドの原因となる。 *3 Zymolyase20Tを加えた時点から細胞壁分解は開始するので、ボルテックス・ミキサー等で強く撹拌してはならない。また、アガロースの温度が高すぎると酵素が失活してしまうので温度には注意する。 *4 この処理により、細胞壁を分解する。 *5 この処理では、SDSによりスフェロプラストを破壊し、プロテアーゼによるタンパク質分解を行う。 *6 各ステップが適切に行われていれば、1年から数年間保存可能である。
1-2. コロニーからPFGEサンプルの調製
2.パルスフィールド電気泳動法による核型解析
適切な電気泳動核型を得るためには、その生物の染色体DNAの大きさによって、分離条件、すなわち、電場を変化させる時間(パルスタイム)と そのときの電場の強さ(電圧)を設定しなければならない。DNAが大きくなるにつれて、パルスタイムを長くし、電圧を低くする必要になる。また、パルスタイムが長くなれば、泳動の時間も長くしなければならない。その泳動条件は経験によるものが大きく、自分のサンプルに応じて設定しなければならないが、すでに、多くの論文で様々な条件が報告されているので、それを参考にするとよい。以下の方法はバイオラッド社製の機械用である。
準備:
10x TBE: 0.9M Tris base、0.9M ホウ酸、40mM EDTA
高ゲル強度アガロース(アガロースHGS、ナカライテスク)
臭化エチジウム
方法:
<〜2Mbまで(1)>
第1条件;120秒パルスタイム、12時間、180V
第2条件;180秒パルスタイム、12時間、180V
<〜2Mbまで(2)>
第1条件;100秒パルスタイム、15時間、180V
第2条件;300秒パルスタイム、20時間、140V
<〜4Mbまで>
第1条件;300秒パルスタイム、24時間、140V
第2条件;1200秒パルスタイム、48時間、80V
- Solutions:
- 1% low melting agarsose
low meting agarose (Wako) --------------- 0.02 g
0.125 M EDTA (pH 7.5) ------------------- 2 ml
- Enzyme soltution
- Zymolyase 20T ----------------------------- 10 mg
SCE -------------------------------------------- 1 ml
- SCE
- 0.1 M Sodium citrate buffer (pH 5.8) ---- 98 ml
0.5 M EDTA (pH 7.5) ------------------------ 2 ml
Sorbitol ---------------------------------------- 18.22 g
# 0.1 M Sodium citrate buffer (pH 5.8)
(A) tri- sodium citrate dihydrate ------ 2.941 g/ 100 ml
(B) citrate ---------------------------------- 2.101 g/ 100 ml
adjust pH to 5.8 by adding (B) to (A).
- LET buffer
- 0.5 M EDTA (pH 7.5) --------------------- 30 ml
1 M Tris- HCl (pH 7.5) -------------------- 1.5 ml
2- mercapoethanol ----------------------- 1.5 ml
- NDS buffer
- 0.5 M EDTA (pH 9.0) --------------------- 30 ml
1 M Tris- HCl (pH 8.3) -------------------- 1.5 ml
SDS ------------------------------------------ 0.018 g
Proteinase K -------------------------------- 5 mg
- 0.5 M EDTA (pH 7.5): per 1 litter
- Na2 EDTA -------------------------------- 186.5 g
NaOH -------------------------------------- 20 g
Adjust pH to 7.5 by adding 5 M NaOH
- 0.5 M EDTA (pH 9.0): per 1 litter
- Na2 EDTA -------------------------------- 186.5 g
NaOH -------------------------------------- 30 g
Adjust pH to 9.0 by adding 5 M NaOH
- 10 X TBE: for 1 litter
- Tris base --------------------------------- 108 g
Boric acid --------------------------------- 55 g
Na2 EDTA -------------------------------- 7.44 g